MARVEL researchers investigate how MOF structures affect dieletric properties
by Carey Sargent, EPFL, NCCR MARVEL
The modern microelectronics industry’s quest to downscale integrated circuits and improve the performance of devices is driving a need for highly efficient electric insulators. The required jump in performance will almost certainly imply moving from traditional silica-based materials to those that feature air gaps—the fundamental idea is to build a structure around the medium featuring the lowest possible dielectric constant, which is vacuum (or air).
Metal–organic frameworks (MOFs), metal ions or clusters coordinated to organic ligands to form one-, two-, or three-dimensional structures, feature large pores and poorly correlated scaffolding—that is the structures are built around air—and so may offer a promising approach. What’s more, they have been studied for more than a couple of decades and can be made with simple, low-cost processes. The class of materials features numerous possible network topologies, enabling the fine-tuning of many properties. Their porous structures and the fact that they are main made up of light atoms such as carbon, hydrogen, nitrogen, and oxygen are linked to the very low mass and electron densities that typically result in low electric susceptibility as well. Rough estimations of the static dielectric constant suggest that MOFs are very promising next-generation interlayer materials.
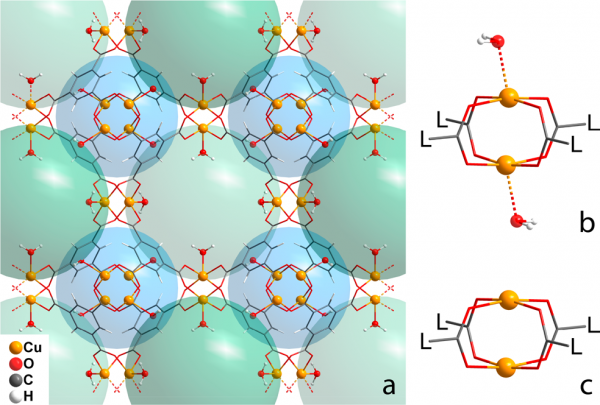
Optimized crystal structure of {Cu3(BTC)2(H2O)3}n: (a)unit cell content, intersecting pores are depicted as cyan and green spheres, (b) paddlewheel with apical ligands, and (c) paddlewheel without apical ligands
Despite this enormous potential, few studies have presented experimental measurements of dielectric constant of MOFs. The results that exist are based on materials that feature different topologies and compositions and refer to different frequency ranges and varying conditions including temperature and atmosphere. The same is true for first-principles simulations. This lack of systematic data of any kind makes it difficult to develop a theory for finding an optimal correlation between structure and properties.
That is, the relationship between a material’s structure and its properties—that a given functionality can be activated or suppressed depending on the nature and arrangement of atoms and molecules as well as the interactions among them—is a central paradigm of material science. At the nanoscale, these actions are governed by quantum mechanics and depend on the distribution and flow of electrons in the field generated by their mutual interplay as well as their interplay with the nuclei. With this in mind, NCCR MARVEL researchers Professor Piero Macchi and PhD student Rebecca Scatena at the University of Bern and Yannick Guntern, now a PhD student at EPFL, started a project to investigate how atomic and electronic structures of MOFs may affect their dielectric properties.
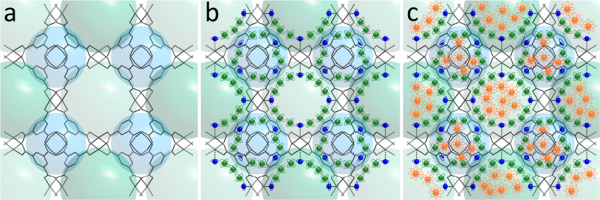
Schematic representation of the porous MOF with different degrees of filling: (a) empty, (b) partially filled, and (c) filled. The vibrational and rotational degrees of freedom increase going in the series: blue, green, and orange.
The researchers selected two isostructural MOFs – {Cu3(BTC)2}n (1, also known as HKUST-1) and {Zn3(BTC)2}n (2), where BTC = 1,3,5-benzenetricarboxylate – for the study because they feature numerous variables that can be changed to reveal their effect on dielectric behavior. The researchers sought to analyze bulk properties over a wider range of frequencies, in particular lower frequencies for potential electronics applications, and to assess key factors that may tune or trigger them. The overall strategy involved investigating the sequence from atomic structure to dielectric function through the electron density distribution and vice versa. Some of the parameters they chose to vary included geometries of the building blocks, connectivity at metal centers, different kinds of sorption on the internal surfaces and in the channels of the MOFs as well as stereo-electronic differences between an open-shell and a closed-shell metal ion.
They managed to pinpoint the contribution of the building blocks to overall macroscopic properties by characterizing bonding properties and estimating the electrostatic forces. Obtaining this information involved quantum crystallographic studies, including the determination of material polarizability from impedance spectroscopy. This then needed to be coupled with first principle simulations to partition the electron density and dielectric polarizability functions in terms of the constituent building blocks.
The experimental determination of accurate electron density distribution in highly porous MOFs is hampered by the poor quality of the crystals, which does not guarantee sufficient X-ray diffraction. To get around this problem, the researchers coupled careful X-ray structural analysis as a function of pore filling with density functional theory (DFT) simulation of the electron charge density distribution in this study.

In the end, the combination of theoretical simulations, X-ray diffraction, thermal gravimetry, and impedance measurements enabled the researchers to draw the following conclusions:
- Empty MOFs are indeed low dielectric constant materials and insensitive to field frequency, mainly because of the large amount of unoccupied and unpolarizable volume inside the framework.
- Guest molecules are easily incorporated in the framework, during the synthesis or exposure of these materials to noninert atmosphere. These parasitic molecules significantly perturb the poorly polarizable volume and enhance the low and high frequency values of the dielectric constant.
- There are three different regimes of sorption, clearly identified by impedance spectroscopy: (1) chemisorption, due to molecules coordinated to the metal paddlewheel SBU, although this interaction remains mostly electrostatic; (2) physisorption, due to molecules hydrogen-bonded to the internal surfaces of the MOFs (thanks to specific binding sites); and (3) mobile molecules in the channels, which are not sorbed on the internal surfaces of the framework.
- The degree of chemisorption also induces some modifications to the framework, especially the coordination at apical free sites of the paddlewheels. Although a strict correlation exists between, for example, M−M distances and the apical coordination, the static dielectric properties of the framework are almost unchanged. This demonstrates the enhanced information on guest binding modes available when the dielectric properties are coupled with the information on the static electric field generated by the MOF.

The researchers believe that the study may encourage further investigations combining X-ray diffraction and impedance spectroscopy. This would allow us to better depict the charge distribution and polarizability inside porous MOFs, therefore identifying the key features of materials that would be suitable for fabricating insulating devices necessary for next-generation microelectronics.
Reference: Electron Density and Dielectric Properties of Highly Porous MOFs:
Binding and Mobility of Guest Molecules in Cu3(BTC)2 and Zn3(BTC)2
Rebecca Scatena,*,† Yannick T. Guntern,†,§ and Piero Macchi*,†,‡
†Department for Chemistry and Biochemistry, University of Bern, Freiestrasse 3, Bern 3012, Switzerland
‡Department of Chemistry, Materials and Chemical Engineering, Polytechnic of Milan, via Mancinelli 7, Milano 20131, Italy
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter